the Peter Kim Lab
Home
Research
Publications
Lab Members
News
Talks
Alumni
Lab Photos
Contact
Home
Research
Publications
Lab Members
News
Talks
Alumni
Lab Photos
Contact
the Peter Kim Lab
Scroll
The Kim lab combines structural biology, protein engineering, immunology, and machine learning to advance global health, with an emphasis on creating vaccines and devising new strategies to enable vaccine generation.
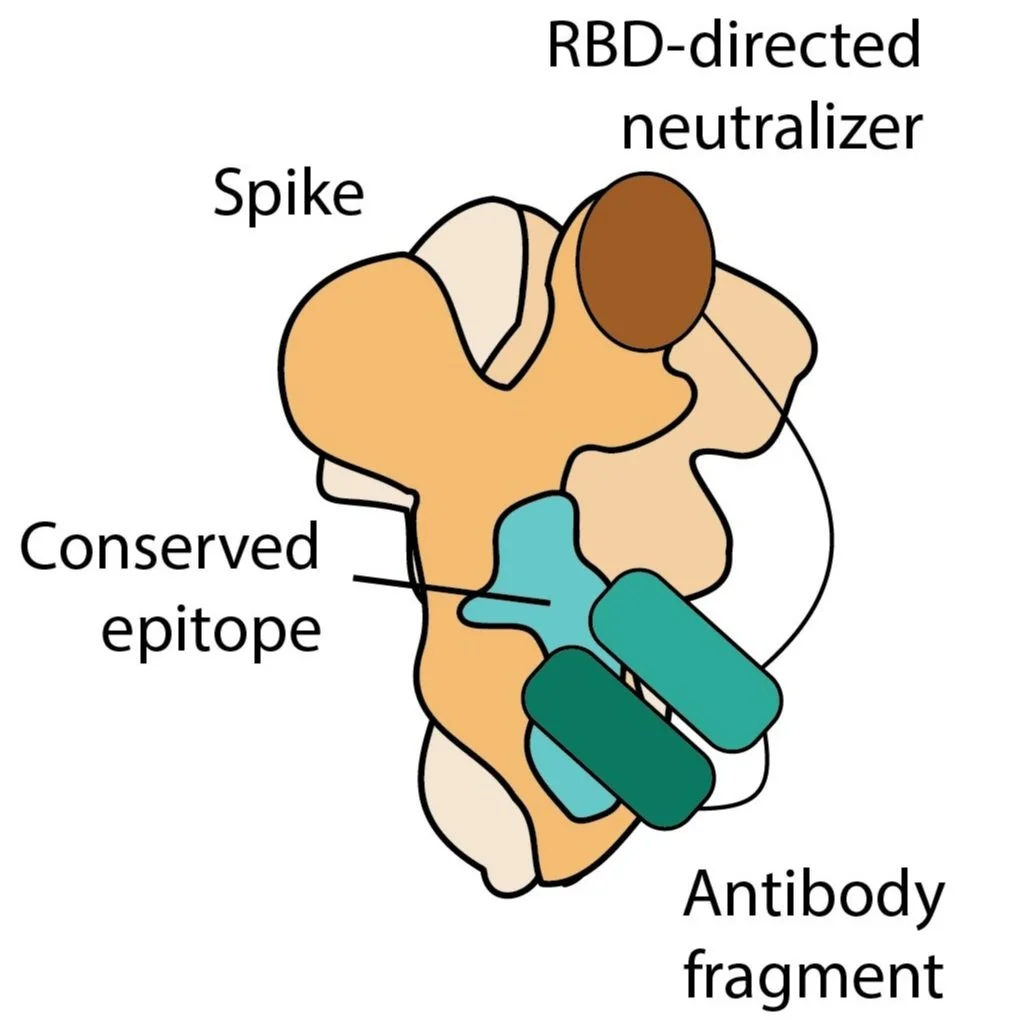
Vaccines and Immunofocusing
ReconnAbs:
receptor-blocking conserved
non-neutralizing antibodies
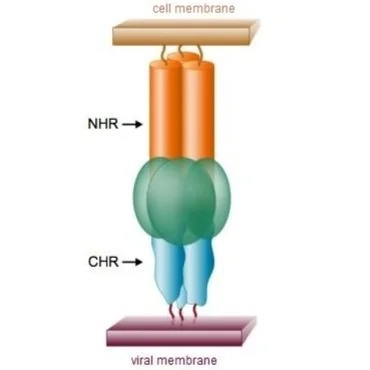
Membrane fusion and the
pre-hairpin intermediate
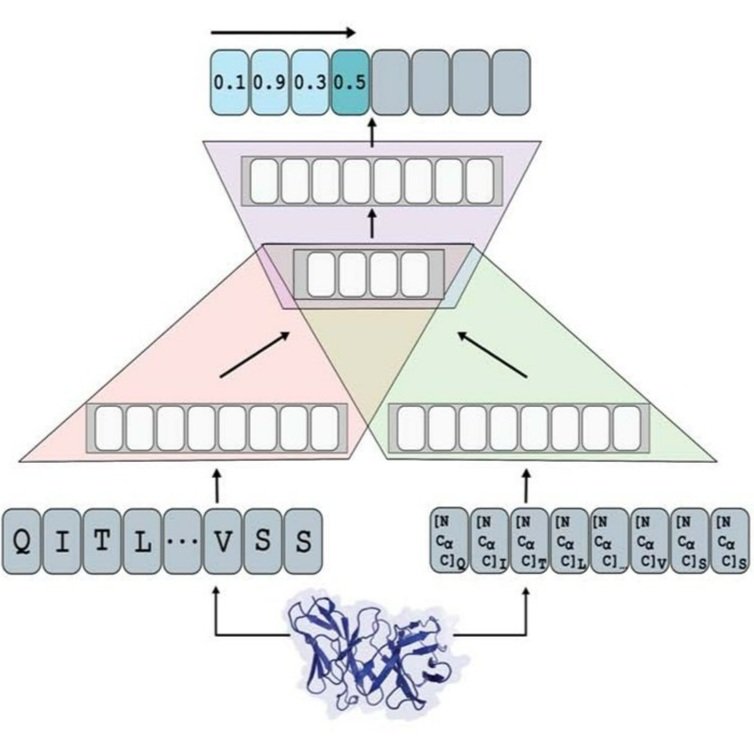
Protein language models for understanding and guiding evolution
Introduction
Summary
Large Image